
Why a physiological environment for mAbs characterization matters?
At Vixen Bio, we believe measuring biologics performance in physiological conditions better predicts their performance or risks in real-world conditions. Next to the specific interactions, additional molecules present in serum will interfere in an (un)specific, but biologically relevant way.
Typically, classic SPR and cell-based assays are used throughout therapeutic development for drug optimization and clinical translation. Understanding specificity and affinity to both the target and potential receptor’s (e.g FcγR), together with recruitment and activation of effector cells is key in selecting best candidates for the wanted therapeutic mode of action. In plasma, serum and the PBMC fraction, many biomolecular interactions can occur due to the rich presence of both plasma (effector) proteins and effector cells, that are initially not assessed during kinetic characterization. In a later stage, traditional functional assays reveal the cytotoxic (or inhibiting?) effect of the therapeutic, but bare little information to the pathway by which and how it was activated, beyond the actual cell-death.
Understanding which molecules are present and how they interact is essential during bio-assay development to ensure the proper data is obtained to guide your further therapeutic development. More importantly, and central to Vixen Bio’s expertise, is the ability to thoughtfully manage these interactions and interpret the data with clarity and confidence. Table 1 describes common serum/plasma compounds and how they could interact in each assay type if not understood.
Table 1: Serum components and how they interact with therapeutic antibodies
| component | conc. / MW / KD | Main interaction with IgG | SPR/BLI | Functional cell assay |
| Complement C1q | ~70–180 µg/mL ~410 kDa subµM KD, extremely avid | Binds clustered Fc on antigen-bound IgG, recognizing CH2. | C1q can bind Fc on surface-captured IgG complexes, increasing apparent mass, altering kinetics, and masking dissociation. | Induce CDC, opsonization, and further complement activation, complicating pure FcγR readouts. |
| Fcγ receptors | Low ng/mL–µg/mL ~30–50 kDa FcγRI: ~1-10 nM KD; FcγRII/III: 0.1-1 µM KD | Bind Fc at CH2–CH3, subclass and glycoform dependent. | Soluble FcγR in serum can bind Fc on immobilized or solution-phase IgG, adding secondary binding signals and apparent affinity shifts. | Effector cell surface FcγR drive ADCC/ ADCP; serum IgG and immune complexes compete for FcγR, alter activation thresholds, introduce donor variability. |
| Complement C3 fragments (C3b, iC3b, C3d) | C3 0.9–1.8 mg/mL C3 ~185 kDa; C3d ~35 kDa 1-50µM KD, high avidity | Covalently attach to immune complexes; interact with complement receptors and sometimes Ig in complexes. | C3 deposition directly on surface or on surface-bound immune complex increases mass. | Drives opsonization and binds complement receptors, adding complement mediated phagocytosis/lysis on top of FcγR or neutralization. |
| Auto-Ab & Anti-drug Ab: Other Ig (e.g. anti-idiotype, RF) | IgG ~0.02–4 µg/mL; IgG ~150 kDa 1-100nM KD | Idiotype, anti-idiotype and rheumatoid factor binding to Fc or Fab. | Polyreactive IgM/IgG or RF in serum can bind the test mAb adding secondary binding phases, apparent avidity, and nonspecific “stickiness”. | RF or anti-idiotypes crosslink therapeutic mAbs, alter effective concentration, trigger off-target activation, or interfere with effector function readouts. |
| Serum albumin (nonspecific, FcRn-linked) | ~35–50 mg/mL ~66 kDa < mM KD | Weak nonspecific binding; cohandled with IgG by FcRn at distinct binding sites. | Can contribute to baseline “matrix effect” and low level nonspecific binding on chips, main impact is bulk refractive index and surface fouling. | Via FcRn co-recycling, albumin can influence IgG trafficking and apparent persistence in long-term cultures or in transcytosis models. |
The complement system, a cornerstone of innate immunity
The complement system is gaining renewed attention as a powerful therapeutic target. This complex cascade of plasma proteins can tag pathogens for destruction, amplify inflammation, and directly lyse cells — functions that are both protective and, when dysregulated, pathogenic. Once considered too intricate for drug development, complement biology is now entering a golden age, driven by better structural understanding and precision pharmacology.
In diseases such as paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS), uncontrolled complement activation destroys red blood cells and tissues, making complement inhibition life-changing. The success of agents targeting C5, like eculizumab and ravulizumab, has validated the pathway clinically and commercially, establishing complement modulation as a proven mechanism of action. Beyond rare diseases, researchers are exploring its role in neurodegeneration, oncology, transplantation, and age-related macular degeneration.
The classical complement pathway is a targeted protein cascade that starts when C1q recognizes antibodies (mainly IgG or IgM) bound to antigens. This triggers a series of cleavages that generate a C3 convertase, which deposits C3b on the target surface and labels it for clearance by phagocytes. As the cascade amplifies, it progresses to C5 cleavage and formation of the membrane attack complex, which can directly damage susceptible cell membranes. Along the way, small fragments such as C3a and C5a are released, acting as inflammatory mediators that recruit and activate immune cells.
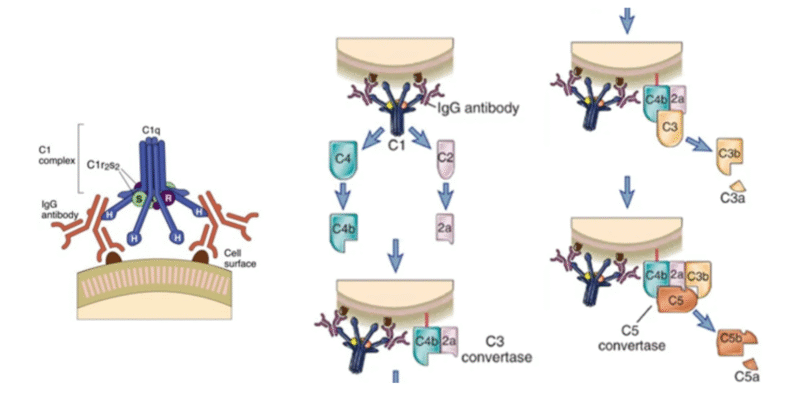
Figure 1: Detail of C1q and description of the complement pathway derived from :Complement Pathways: Types, Functions, Regulation • Microbe Online.
Kinetics in buffer versus serum
In this case study, we wanted to investigate both the target binding and the physiological functional potency of a monoclonal antibody (mAb) candidate developed against a target to treat breast cancer. We compared the mAb candidate binding kinetics to its target in buffer and (undiluted) human serum. This showed dramatic differences in curve shapes and levels of signal.
Kinetics of an antibody antigen interaction can be dramatically changed by measuring it in serum vs buffer. In app note “Leading to patient-dependent affinity” Vixen Bio shows that a similar affinity in buffer of IFX and a biosimilar to TNF changes to a factor 10 difference in plasma.
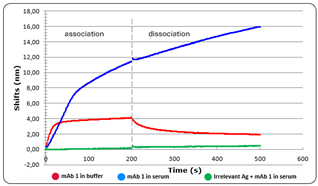
Figure 2: mAb 1 in buffer (red) versus serum (blue) compared to irrelevant Ag in the same serum (green).
In this figure a dramatic increase of binding triggered by the mAb Is seen with a signal roughly 3 times higher in serum for the considered association time. Interestingly, the signal kept on increasing even during the dissociation step in serum suggesting other compounds with either low affinity and high concentration or avidity sensitive molecules such as complement proteins may keep binding to antigen-mAb complexes.
Heat Inactivation removes complement interactions
For practical assay design, as heat-inactivation of the serum destroys the complement proteins the clean antigen–antibody interaction becomes visible in the kinetic data. As the sample is also free of FcγR/FcRn proteins, measurement in buffer vs. native serum allows to quantify the magnitude of the serum effects. As such, we spiked the mAb candidate in heat-inactivated human serum and compared the association of the mAb candidate to its target in native vs. heat-inactivated serum. The candidate kinetics binding in heat-inactivated serum was similar to that obtained in buffer.
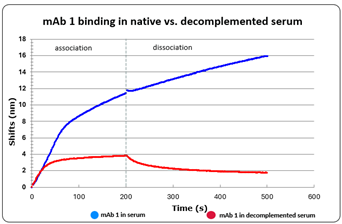
Figure 3: mAb 1 kinetics in serum (blue) versus heat inactivated serum (red)
When focusing on the association step, the difference between the mAb binding in native and heat-inactivated serum allows to visualize the complement binding in serum. This unique ability allows to compare mAb candidates not only on their affinity for the target but also on their Fc potency to recruit the complement, thus making the candidate selection both more physiologically relevant and more robust.
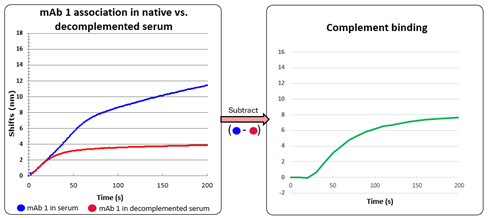
Figure 4: zoom in on the association of mAb1 in serum (blue) and decomplemented serum(red) (left). Subtracted association isolating the non mAb1 signal of serum binding (green) (right).
Is it the complement pathway?
The reasoning for the complement pathway identification is that C1q binds very quickly (seconds) to hexamer arranged Fc tails of the IgG. Sufficient density on the sensor probe for the hexamer of IgG to form is achieved after about 25s resulting in a quick uptake of additional mass. The C1 complex is about 760 kDa or 130 kDa per IgG bound on the surface. As the mass dependent signal generated by the serum is more than double the isolated mAb1 association, this strengthens the proof of additional interactions of e.g. C4b2a with the sensor probe directly. These signals even pertain when no extra antibody is binding at the plateaued association and even during dissociation. These kinetic signatures do not match with Fcy receptors, other IgG’s or albumin behavior.
Heat-inactivated sera contain reduced amounts of complement factors (C1–C9) and regulators like C1 inhibitor, while leaving immunoglobulins largely intact, but with increased concentrations of circulating immune complexes.1 Idiotypic IgG’s are still present in heat inactivated serum. The sensorgrams shown here in comparing native and heat inactivated serum proof the minimal influence of autoantibodies or anti-drug antibodies. Therefore, the absence of a signal in the heat-inactivated sensorgram (Figure 4) indicates that the observed effect is most likely due to complement interference rather than other immune mechanisms described in the table above.
Conclusion
Testing a therapeutic candidate in serum can confirm the wanted mode of action or flag an unwanted effect and is as such a measure for serum functionality. In the shown method it allows to help discriminate the different types of interactions with immune system components (complement, autoantibodies, Fc receptors). Furthermore, it can help verify the correct pathway activation, otherwise obscured in blind cell-based assays, such as for CDC (complement dependent cytotoxicity). This type of data strengthens the submission files of new therapeutic molecules after development and confirm whether the mode of action upholds in a diverse patient population. In this use case we were able to flag the higher than expected complement activation even before animal model testing. The results urged the customer to re-engineer their main candidate with low Fc engagement mutations.
We’re sharing this summary to spotlight key developments shaping the field using Vixen Bio technology. Interested in learning how our physiological assays can help streamline your therapeutic development and derisk preclinically?
Let’s connect and discuss how we can support your work.
Interested in more details? info@vixenbio.com or follow us on Linkedin
References
- Fante, M.; Decking, S.-M.; Bruss, C.; Schreml, S.; Siska, P.; Kreutz, M.; Renner, K. (2021). Heat-Inactivation of Human Serum Destroys C1 Inhibitor, Pro-motes Immune Complex Formation, and Improves Human T Cell Function. International Journal of Molecular Sciences. 22. 2646. 10.3390/ijms22052646.